
Thermodynamics and kinetics of the amyloid-β peptide revealed by Markov state models based on MD data in agreement with experiment†

Abstract
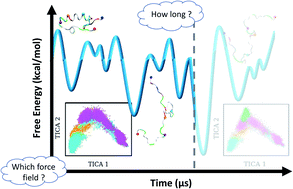
The amlyoid-β peptide (Aβ) is closely linked to the development of Alzheimer's disease. Molecular dynamics (MD) simulations have become an indispensable tool for studying the behavior of this peptide at the atomistic level. General key aspects of MD simulations are the force field used for modeling the peptide and its environment, which is important for accurate modeling of the system of interest, and the length of the simulations, which determines whether or not equilibrium is reached. In this study we address these points by analyzing 30-μs MD simulations acquired for Aβ40 using seven different force fields. We assess the convergence of these simulations based on the convergence of various structural properties and of NMR and fluorescence spectroscopic observables. Moreover, we calculate Markov state models for the different MD simulations, which provide an unprecedented view of the thermodynamics and kinetics of the amyloid-β peptide. This further allows us to provide answers for pertinent questions, like: which force fields are suitable for modeling Aβ? (a99SB-UCB and a99SB-ILDN/TIP4P-D); what does Aβ peptide really look like? (mostly extended and disordered) and; how long does it take MD simulations of Aβ to attain equilibrium? (at least 20–30 μs). We believe the analyses presented in this study will provide a useful reference guide for important questions relating to the structure and dynamics of Aβ in particular, and by extension other similar disordered proteins.
 Please wait while we load your content...
Please wait while we load your content...

