
Dispersion forces drive water oxidation in molecular ruthenium catalysts†

Abstract
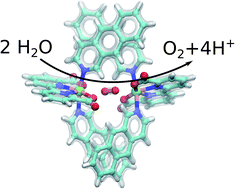
Rational design of artificial water-splitting catalysts is central for developing new sustainable energy technology. However, the catalytic efficiency of the natural light-driven water-splitting enzyme, photosystem II, has been remarkably difficult to achieve artificially. Here we study the molecular mechanism of ruthenium-based molecular catalysts by integrating quantum chemical calculations with inorganic synthesis and functional studies. By employing correlated ab initio calculations, we show that the thermodynamic driving force for the catalysis is obtained by modulation of π-stacking dispersion interactions within the catalytically active dimer core, supporting recently suggested mechanistic principles of Ru-based water-splitting catalysts. The dioxygen bond forms in a semi-concerted radical coupling mechanism, similar to the suggested water-splitting mechanism in photosystem II. By rationally tuning the dispersion effects, we design a new catalyst with a low activation barrier for the water-splitting. The catalytic principles are probed by synthesis, structural, and electrochemical characterization of the new catalyst, supporting enhanced water-splitting activity under the examined conditions. Our combined findings show that modulation of dispersive interactions provides a rational catalyst design principle for controlling challenging chemistries.
 Please wait while we load your content...
Please wait while we load your content...

