
B12X11(H2)−: exploring the limits of isotopologue selectivity of hydrogen adsorption†

Abstract
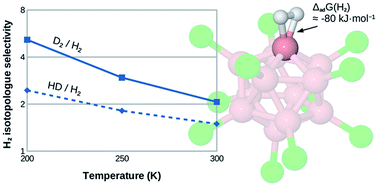
We study the isotopologue-selective binding of dihydrogen at the undercoordinated boron site of B12X11− (X = H, F, Cl, Br, I, CN) using ab initio quantum chemistry. With a Gibbs free energy of H2 attachment reaching up to 80 kJ mol−1 (ΔG at 300 K for X = CN), these sites are even more attractive than most undercoordinated metal centers studied so far. We thus believe that they can serve as an edge case close to the upper limit of isotopologue-selective H2 adsorption sites. Differences of the zero-point energy of attachment average 5.0 kJ mol−1 between D2 and H2 and 2.7 kJ mol−1 between HD and H2, resulting in hypothetical isotopologue selectivities as high as 2.0 and 1.5, respectively, even at 300 K. Interestingly, even though attachment energies vary substantially according to the chemical nature of X, isotopologue selectivities remain very similar. We find that the H–H activation is so strong that it likely results in the instantaneous heterolytic dissociation of H2 in all cases (except, possibly, for X = H), highlighting the extremely electrophilic nature of B12X11− despite its negative charge. Unfortunately, this high reactivity also makes B12X11− unsuitable for practical application in the field of dihydrogen isotopologue separation. Thus, this example stresses the two-edged nature of strong H2 affinity, yielding a higher isotopologue selectivity on the one hand but risking dissociation on the other, and helps define a window of adsorption energies into which a material for selective adsorption near room temperature should ideally fall.
 Please wait while we load your content...
Please wait while we load your content...

