
Geometry transformation of ionic surfactants and adsorption behavior on water/n-decane-interface: calculation by molecular dynamics simulation and DFT study†
 *abc
and
Fang
Yu
*bc
*abc
and
Fang
Yu
*bc
Abstract
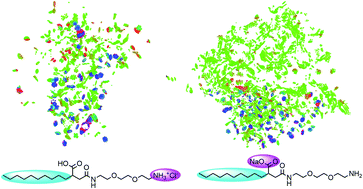
Understanding the effect of surfactant structure on their ability to modify interfacial properties is of great scientific and industrial interest. In this work, we have synthesized four amide based ionic surfactants under acidic or basic conditions, including CTHA·HCl, CTEA·HCl, CTHA−Na+ and CTEA−Na+. Experiments have proved that the anionic surfactant with polyethylene oxide groups (CTEA−Na+) had the lowest surface tension on the water/n-decane interface. Molecular dynamics simulations have been applied to investigate the structural effect on the adsorption behavior of four different surfactants. The surface tension, interface thickness, interface formation energy, density profiles, order parameters, radial distribution function on the water/n-decane interfaces were calculated and compared. During the equilibrium states, we found that the interface configuration of two cationic surfactants are almost linear while the two anionic surfactants are changed to bending shapes due to the different positions of the hydrophilic head groups. Further DFT study and wavefunction analysis of surfactants have shown that CTEA−Na+ can form stronger vdW interactions with n-decane molecules due to a more neutral electrostatic potential distribution. Meanwhile, the introduction of polyethylene oxide groups has offered more H-bonding sites and resulted in more concentrated H-bonding interactions with water molecules. The difference of weak interactions may contribute to the conformational change and finally affect the interface properties of these ionic surfactants.
 Please wait while we load your content...
Please wait while we load your content...

