
Elucidating the intercalation of methylated 1,10-phenanthroline with DNA: the important weight of the CH/H interactions and the selectivity of CH/π and CH/n interactions†
 *ab
*ab
Abstract
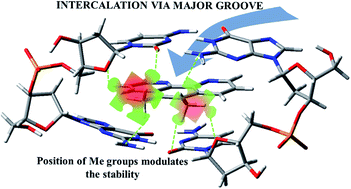
Flat molecules like phenanthroline derivatives intercalate between base pairs of deoxyribonucleic acid and produce cytotoxic effects against tumoral cells. Elucidating the way of intercalation and its modulation on their efficiency by substitution still remains a challenging topic of research. In this work we analysed the intercalation via the major groove of methylated derivatives of phenanthroline, in different number and position, between guanine–cytosine base pairs. We studied our systems by using semi-empirical methods and density functional theory including dispersion corrections with the PM6-DH2 Hamiltonian and the B3LYP-D3 functional. We explored the geometry and electronic structure by means of the quantum theory of atoms in molecules and non-covalent interactions index analyses, whereas the interaction energy was estimated by means of two different approaches: one taking into account the results from the quantum theory of atoms in molecules analysis and the other based on the so-called energy decomposition analysis. The effect of solvation was also taken into consideration. Our studies show that CH/π and CH/n interactions by means of the –CH3 groups of methylated phen follow a clear pattern for any number of –CH3 groups and their position in the methylated phen ligand. That is, they try to produce the CH/π and CH/n interactions with the O and N heteroatoms of the base pairs and with the O atoms of the sugar and phosphate backbone. These findings suggest that the modulation of the intercalation of ligands that are able to form CH/π and CH/n weak interactions with the deoxyribonucleic acid is ruled not only by the number and position of the substitutions of the ligands but also by some key sites, which are the O and N atoms of the deoxyribonucleic acid in our analysed systems. It suggests some key and lock mechanism in which the interacting fragments fit like puzzle pieces in order to achieve the optimal interaction for the stabilization of the system. Interaction energies were calculated by using different approaches which converged to similar trends about the number and position of the –CH3 groups. The important weight of the CH/H interactions in the total interaction energy must be highlighted.
 Please wait while we load your content...
Please wait while we load your content...

