
Inhibition mechanism and hot-spot prediction of nine potential drugs for SARS-CoV-2 Mpro by large-scale molecular dynamic simulations combined with accurate binding free energy calculations†

Abstract
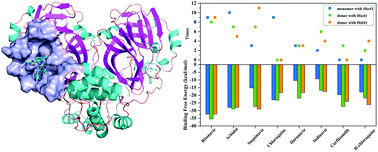
Coronavirus disease 2019 (COVID-19), which is caused by a new coronavirus known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is spreading around the world. However, a universally effective treatment regimen has not been developed to date. The main protease (Mpro), a key enzyme of SARS-CoV-2, plays a crucial role in the replication and transcription of this virus in cells and has become the ideal target for rational antiviral drug design. In this study, we performed molecular dynamics simulations three times for these complexes of Mpro (monomeric and dimeric) and nine potential drugs that have a certain effect on the treatment of COVID-19 to explore their binding mechanism. In addition, a total of 12 methods for calculating binding free energy were employed to determine the optimal drug. Ritonavir, Arbidol, and Chloroquine consistently showed an outstanding binding ability to monomeric Mpro under various methods. Ritonavir, Arbidol, and Saquinavir presented the best performance when binding to a dimer, which was independent of the protonated state of Hie41 (protonated at Nε) and Hid41 (protonated at Nδ), and these findings suggest that Chloroquine may not effectively inhibit the activity of dimeric Mproin vivo. Furthermore, three common hot-spot residues of Met165, Hie41, and Gln189 of monomeric Mpro systems dominated the binding of Ritonavir, Arbidol, and Chloroquine. In dimeric Mpro, Gln189, Met165, and Met49 contributed significantly to binding with Ritonavir, Arbidol, and Saquinavir; therefore, Gln189 and Met165 might serve as the focus in the discovery and development of anti-COVID-19 drugs. In addition, the van der Waals interaction played a significant role in the binding process, and the benzene ring of the drugs showed an apparent inhibitory effect on the normal function of Mpro. The binding cavity had great flexibility to accommodate these different drugs. The results would be notably helpful for enabling a detailed understanding of the binding mechanisms for these important drug–Mpro interactions and provide valuable guidance for the design of potent inhibitors.
 Please wait while we load your content...
Please wait while we load your content...

