
An integrated network analysis approach to identify potential key genes, transcription factors, and microRNAs regulating human hematopoietic stem cell aging†
 a
and
Manoj
Kumar
*ab
a
and
Manoj
Kumar
*ab
Abstract
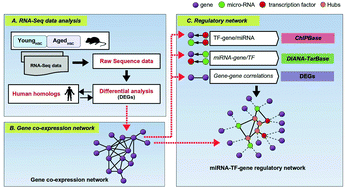
Hematopoietic stem cells (HSCs) undergo functional deterioration with increasing age that causes loss of their self-renewal and regenerative potential. Despite various efforts, significant success in identifying molecular regulators of HSC aging has not been achieved, one prime reason being the non-availability of appropriate human HSC samples. To demonstrate the scope of integrating and re-analyzing the HSC transcriptomics data available, we used existing tools and databases to structure a sequential data analysis pipeline to predict potential candidate genes, transcription factors, and microRNAs simultaneously. This sequential approach comprises (i) collecting matched young and aged mice HSC sample datasets, (ii) identifying differentially expressed genes, (iii) identifying human homologs of differentially expressed genes, (iv) inferring gene co-expression network modules, and (v) inferring the microRNA–transcription factor–gene regulatory network. Systems-level analyses of HSC interaction networks provided various insights based on which several candidates were predicted. For example, 16 HSC aging-related candidate genes were predicted (e.g., CD38, BRCA1, AGTR1, GSTM1, etc.) from GCN analysis. Following this, the shortest path distance-based analyses of the regulatory network predicted several novel candidate miRNAs and TFs. Among these, miR-124-3p was a common regulator in candidate gene modules, while TFs MYC and SP1 were identified to regulate various candidate genes. Based on the regulatory interactions among candidate genes, TFs, and miRNAs, a potential regulation model of biological processes in each of the candidate modules was predicted, which provided systems-level insights into the molecular complexity of each module to regulate HSC aging.
 Please wait while we load your content...
Please wait while we load your content...