
Photodissociation dynamics of halogenated aromatic molecules: the case of core-ionized tetrabromothiophene†

Abstract
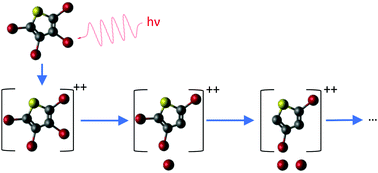
We studied the gas-phase photodissociation of a fully halogenated aromatic molecule, tetrabromothiophene, upon core–shell ionization by using synchrotron radiation and energy-resolved multiparticle coincidence spectroscopy. Photodynamics was initiated by the selective soft X-ray ionization of three elements – C, S, and Br – leading to the formation of dicationic states by Auger decay. From a detailed study of photodissociation upon Br 3d ionization, we formulate a general fragmentation scheme, where dissociation into neutral fragments and a pair of cations prevails, but dicationic species are also produced. We conclude that dicationic tetrabromothiophene typically undergoes deferred charge separation (with one of the ions being often Br+) that may be followed by secondary dissociation steps, depending on the available internal energy of the parent dication. Observations suggest that the ejection of neutral bromine atoms as the first step of deferred charge separation is a prevailing feature in dicationic dissociation, although sometimes in this step the C–Br bonds appear to remain intact and the thiophene ring is broken instead. Ionization-site-specific effects are observed particularly in doubly charged fragments and as large differences in the yields of the intact parent dication. We interpret these effects, using first-principles calculations and molecular dynamics simulations of core-hole states, as likely caused by the geometry changes during the core-hole lifetime.
 Please wait while we load your content...
Please wait while we load your content...

