
Dipolar repulsion in α-halocarbonyl compounds revisited†
 ab
Lucas
de Azevedo Santos,
ab
Trevor A.
Hamlin,
a
F. Matthias
Bickelhaupt,
*ac
Matheus
P. Freitas
*b
and
Célia
Fonseca Guerra
*ad
ab
Lucas
de Azevedo Santos,
ab
Trevor A.
Hamlin,
a
F. Matthias
Bickelhaupt,
*ac
Matheus
P. Freitas
*b
and
Célia
Fonseca Guerra
*ad
Abstract
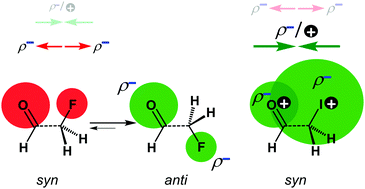
The concept of dipolar repulsion has been widely used to explain several phenomena in organic chemistry, including the conformational preferences of carbonyl compounds. This model, in which atoms and bonds are viewed as point charges and dipole moment vectors, respectively, is however oversimplified. To provide a causal model rooted in quantitative molecular orbital theory, we have analyzed the rotational isomerism of haloacetaldehydes OHC–CH2X (X = F, Cl, Br, I), using relativistic density functional theory. We have found that the overall trend in the rotational energy profiles is set by the combined effects of Pauli repulsion (introducing a barrier around gauche that separates minima at syn and anti), orbital interactions (which can pull the anti minimum towards anticlinal to maximize hyperconjugation), and electrostatic interactions. Only for X = F, not for X = Cl–I, electrostatic interactions push the preference from syn to anti. Our bonding analyses show how this trend is related to the compact nature of F versus the more diffuse nature of the heavier halogens.
- This article is part of the themed collection: 2021 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

