
Determining usefulness of machine learning in materials discovery using simulated research landscapes†
 *a
and
Alessandro
Troisi
*a
*a
and
Alessandro
Troisi
*a
Abstract
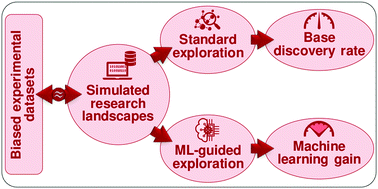
When existing experimental data are combined with machine learning (ML) to predict the performance of new materials, the data acquisition bias determines ML usefulness and the prediction accuracy. In this context, the following two conditions are highly common: (i) constructing new unbiased data sets is too expensive and the global knowledge effectively does not change by performing a limited number of novel measurements; (ii) the performance of the material depends on a limited number of physical parameters, much smaller than the range of variables that can be changed, albeit such parameters are unknown or not measurable. To determine the usefulness of ML under these conditions, we introduce the concept of simulated research landscapes, which describe how datasets of arbitrary complexity evolve over time. Simulated research landscapes allow us to use different discovery strategies to compare standard materials exploration with ML-guided explorations, i.e. we can measure quantitatively the benefit of using a specific ML model. We show that there is a window of opportunity to obtain a significant benefit from ML-guided strategies. The adoption of ML can take place too soon (not enough information to find patterns) or too late (dense datasets only allow for negligible ML benefit), and the adoption of ML can even slow down the discovery process in some cases. We offer a qualitative guide on when ML can accelerate the discovery of new best-performing materials in a field under specific conditions. The answer in each case depends on factors like data dimensionality, corrugation and data collection strategy. We consider how these factors may affect the ML prediction capabilities and discuss some general trends.
- This article is part of the themed collections: 2021 PCCP HOT Articles and Emerging AI Approaches in Physical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

