
Polycyclic aromatic hydrocarbons: from small molecules through nano-sized species towards bulk graphene†
 *a
and
Amir
Karton
b
*a
and
Amir
Karton
b
Abstract
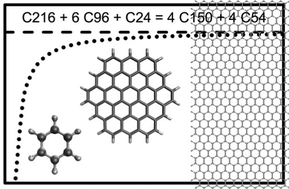
We have examined the use of systematic bond-separation reactions and purposely constructed chemistry-preserving isodesmic reactions for the thermochemical calculation of aromatic hydrocarbon species. The bond-separation approach yields somewhat disappointing accuracy even when the reaction energies are obtained with generally robust composite and double-hybrid (DH) density functional theory (DFT) methods. In contrast, for the purposely constructed reactions, we find a dramatic improvement in the accuracy for energies calculated with all methods examined. Notably, for medium-sized aromatic hydrocarbons, we find that an effective approach for formulating a well-balanced reaction is to split the target species into two halves with an aromatic overlapping region. Overall, the G4(MP2)-XK, MPW2PLYP, MN15, PBE, and DC-DFTB3 methods are reasonable within their respective classes of methods for the calculation of bond-separation as well as chemistry-preserving isodesmic reactions. We have further computed per-carbon atomization energy (AE) for a series of D6h benzene-type molecules, and thus obtained a formula for extrapolation to the graphene limit [AEn = 711.5 × (1 − 1/n0.640) kJ mol−1, where n = number of carbons]. It suggests that nano-graphene with a length larger than 10 nm would resemble properties of bulk graphene, and conversely, downsizing a nano-graphene beyond this point may lead to considerably altered properties from the bulk.
 Please wait while we load your content...
Please wait while we load your content...

