
Doping of the hydrogen-passivated Si(100) electronic structure through carborane adsorption studied using density functional theory†

Abstract
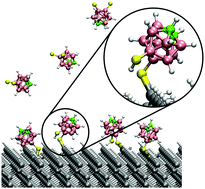
Adsorption of molecular materials with tailored chemical properties represents a new and promising avenue to non-destructively dope silicon. Dithiocarboranes possess large permanent dipoles and readily form stable monolayers on a variety of substrates. Here we use density functional theory to investigate the doping of hydrogen-passivated Si(100) substrates through the adsorption of dithiocarborane molecules. We find that dithiocarboranes can both physisorb and chemisorb on the substrate. Chemisorbed structures arise when a S atom in the molecular linker group replaces a surface H atom. We establish the formation of these Si-molecule bonds and characterize their mechanical and thermal stability. Analysis of the calculated electronic structure of adsorbed interfaces shows that carborane adsorption does not result in interface gap states. The band gap in adsorbed junctions is defined by Si states and its magnitude is almost unchanged with respect to the clean Si slab. The large carborane electrostatic dipole results in the downwards shift of Si spectral features by 0.3 eV, reducing the hole injection barrier by this amount with respect to the pristine Si substrate. Molecular dynamics simulations reveal these structural and electronic features to be stable at room temperature. Our work shows that molecular adsorbates having large electrostatic dipoles are a promising strategy to non-destructively dope semiconductor substrates.
 Please wait while we load your content...
Please wait while we load your content...

