
Acenes and phenacenes in their lowest-lying triplet states. Does kinked remain more stable than straight?†

Abstract
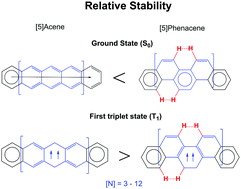
The larger stability of phenacenes compared to their acene isomers in their ground states is attributed to the larger aromaticity of the former. To our knowledge the relative stability of acenes and phenacenes in their lowest-lying triplet states (T1) has not been discussed yet. Using unrestricted density functional theory calculations, our results show that for the smallest members of the series, acenes in their T1 states are more stable than the corresponding phenacenes. However, when the number of the rings (n) involved increases, the energy difference is reduced and for n > 12, phenacenes become more stable than acenes in their T1 states. To rationalize this trend, we analyze the aromaticity of acenes and phenacenes using a set of aromaticity descriptors. We find that in the T1 states of both acenes and phenacenes, the outer rings form aromatic Clar π-sextets. In acenes, delocalization of spin density in the central rings leads to the preferred formation of the largest antiaromatic diradical. Resonant structures in the form of antiaromatic diradical Baird π-octadectets and π-tetradectets are the major contributors, while the smaller ones, such as π-doublets and π-sextets, contribute the least. In phenacenes, structures with diradical antiaromatic Baird π-sextets in some of the central rings contribute the most. These results are relevant to understand the (anti)aromaticity of larger polycyclic aromatic hydrocarbons in their triplet states.
 Please wait while we load your content...
Please wait while we load your content...

