
The investigation of methane storage at the Ni-MOF-74 material: a periodic DFT calculation†

Abstract
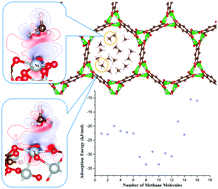
To develop a high-performance methane storage material, an understanding of the mechanism and electronic interactions between methane and the material is essential. In this study, we performed detailed theoretical analyses to investigate the methane storage capacity of Ni-MOF-74 using a large-scale periodic DFT code CONQUEST. In a single pore of the unit cell, we considered three possible sites, iSBU, L, and P sites, where iSBU is the inorganic secondary building unit with a metal center, and L is the linker consisting of the organic building unit, while the P site is the vacuum site in the center of the pore. It shows that the methane molecule adsorption possesses the largest methane molecule adsorption energy on the iSBU site. Our calculations indicate that both C–H⋯O and weak agostic interactions exist between the methane molecule and the iSBU site. The adsorption energy of one methane molecule on the iSBU site is in good agreement with previous experimental and theoretical studies. The calculation of the stepwise methane molecule adsorption shows that the first six methane molecules can first occupy the iSBU sites via C–H⋯O and weak agostic interactions. The second six methane molecules are adsorbed on the remaining L sites, where the C–H⋯π interaction becomes important, leading to the synergistic effect together with the C–H⋯O interaction to enhance the adsorption energy of the methane molecule. Finally, it can adsorb up to sixteen CH4 molecules in a single pore of a unit cell at Ni-MOF-74. Moreover, we conducted DOS and EDD analyses, which clearly show that the interactions play a vital role in the adsorption of a methane molecule on Ni-MOF-74, especially the C–H⋯O interaction.
 Please wait while we load your content...
Please wait while we load your content...

