
Ab initio screening of Pt-based transition-metal nanoalloys using descriptors derived from the adsorption and activation of CO2†

Abstract
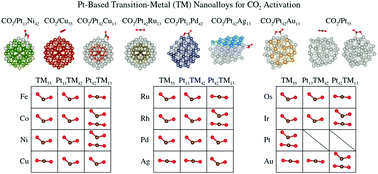
In this study, we report an ab initio screening, based on density functional theory calculations, of Pt-based transition-metal nanoalloys using physicochemical descriptors derived from the adsorption and activation of CO2 on 55-atom nanoclusters, namely, PtnTM55−n, with n = 0, 13, 42, 55, TM = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Au. From the adsorption on the unary and binary nanoclusters, at the chemisorption regime (bent CO2), we identified a linear correlation between the interaction energy and charge transfer from the nanoclusters towards CO2 and the bent CO2 angle; moreover, the interaction energy is enhanced for larger values of the molecular charge and angle. The alloying of Cu55, Ag55, and Au55 with Pt provides a path to change the CO2 adsorption from physisorption (linear, non-activated) to chemisorption (enhanced interaction energies, bent, activated), while the strong interaction energy of CO2 with Os55, Ru55, and Fe55 can be decreased by alloying with Pt using different structural configurations, i.e., the trends are similar for core–shell and segregated structures. Thus, based on our results and analyses, we can select different combinations of PtnTM55−n nanoalloys to yield the desired interaction strength and magnitude of the charge transfer towards the activated anionic CO2, which can help in the design of nanocatalysts for CO2 activation or different chemical reactions in which charge transfer plays a crucial role.
 Please wait while we load your content...
Please wait while we load your content...

