
Predicting OH stretching fundamental wavenumbers of alcohols for conformational assignment: different correction patterns for density functional and wave-function-based methods†
 *a
and
Martin A.
Suhm
a
*a
and
Martin A.
Suhm
a
Abstract
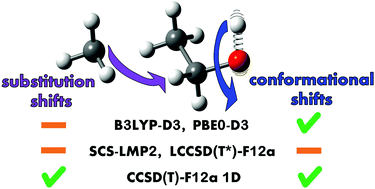
A model is presented for the prediction of OH stretching fundamental wavenumbers of alcohol conformers in the gas phase by application of a small set of empirical anharmonicity corrections to calculations in the harmonic approximation. In contrast to the popular application of a uniform scaling factor, the local chemical structure of the alcohol is taken into account to greatly improve accuracy. Interestingly, different correction patterns emerge for results of hybrid density functional (B3LYP-D3 and PBE0-D3) and wave-function-based methods (SCS-LMP2, LCCSD(T*)-F12a and CCSD(T)-F12a 1D). This raises questions about electronic structure deficiencies in these methods and differences in anharmonicity between alcohols. After its initial construction on the basis of literature assignments the model is tested with Raman jet spectroscopy of propargyl alcohol, cyclohexanol, borneol, isopinocampheol and 2-methylbutan-2-ol. For propargyl alcohol a spectral splitting attributed to tunneling is resolved. PBE0-D3 is identified as a well performing and broadly affordable electronic structure method for this model. A mean absolute error of 1.3 cm−1 and a maximum absolute error of 3 cm−1 result for 46 conformers of 24 alcohols in a 60 cm−1 range, when a single parameter is adjusted separately for each alcohol substitution class (methanol, primary, secondary, tertiary).
 Please wait while we load your content...
Please wait while we load your content...

