
Interplay of physically different properties leading to challenges in separating lanthanide cations – an ab initio molecular dynamics and experimental study†
 a
and
a
and
Abstract
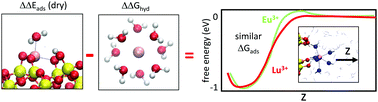
Lanthanide elements have well-documented similarities in their chemical behavior, which make the valuable trivalent lanthanide cations (Ln3+) particularly difficult to separate from each other in water. In this work, we apply ab initio molecular dynamics simulations to compare the free energies (ΔGads) associated with the adsorption of lanthanide cations to silica surfaces at a pH condition where SiO− groups are present. The predicted ΔGads for lutetium (Lu3+) and europium (Eu3+) are similar within statistical uncertainties; this is in qualitative agreement with our batch adsorption measurements on silica. This finding is remarkable because the two cations exhibit hydration free energies (ΔGhyd) that differ by >2 eV, different hydration numbers, and different hydrolysis behavior far from silica surfaces. We observe that the similarity in Lu3+ and Eu3+ ΔGads is the result of a delicate cancellation between the difference in Eu3+ and Lu3+ hydration (ΔGhyd), and their difference in binding energies to silica. We propose that disrupting this cancellation at the two end points, either for adsorbed or completely desorbed lanthanides (e.g., via nanoconfinment or mixed solvents), will lead to effective Ln3+ separation.
 Please wait while we load your content...
Please wait while we load your content...

