
Reducing crystal structure overprediction of ibuprofen with large scale molecular dynamics simulations†

Abstract
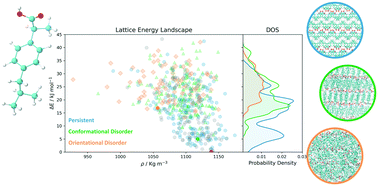
The control of the crystal form is a central issue in the pharmaceutical industry. The identification of putative polymorphs through Crystal Structure Prediction (CSP) methods is based on lattice energy calculations, which are known to significantly over-predict the number of plausible crystal structures. A valuable tool to reduce overprediction is to employ physics-based, dynamic simulations to coalesce lattice energy minima separated by small barriers into a smaller number of more stable geometries once thermal effects are introduced. Molecular dynamics simulations and enhanced sampling methods can be employed in this context to simulate crystal structures at finite temperature and pressure. Here we demonstrate the applicability of approaches based on molecular dynamics to systematically process realistic CSP datasets containing several hundreds of crystal structures. The system investigated is ibuprofen, a conformationally flexible active pharmaceutical ingredient that crystallises both in enantiopure forms and as a racemic mixture. By introducing a hierarchical approach in the analysis of finite-temperature supercell configurations, we can post-process a dataset of 555 crystal structures, identifying 65% of the initial structures as labile, while maintaining all the experimentally known crystal structures in the final, reduced set. Moreover, the extensive nature of the initial dataset allows one to gain quantitative insight into the persistence and the propensity to transform of crystal structures containing common hydrogen-bonded intermolecular interaction motifs.
- This article is part of the themed collection: Computer Aided Solid Form Design
 Please wait while we load your content...
Please wait while we load your content...

