
On the understanding of the optoelectronic properties of S-doped MoO3 and O-doped MoS2 bulk systems: a DFT perspective†
 *ab
and
Tangui
Le Bahers
*a
*ab
and
Tangui
Le Bahers
*a
Abstract
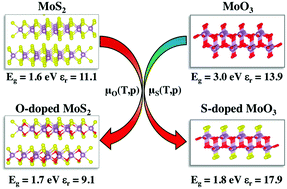
First-principles calculations are carried out to understand the structure and optoelectronic properties of α-MoO3 and 2H-MoS2 bulk systems with anionic isovalent-atom substitutions. Density functional theory (DFT) calculations are performed by adopting the HSE06 functional to probe the optoelectronic structures and the Boltzmann transport theory to compute the DOS-averaged effective mass and mobility of the studied compounds. We show that substituting oxygen atoms by sulfur atoms in α-MoO3, the electronic energy gap is decreased from 3.0 eV in the pristine material to around 1.6 eV in the sulfur doped material. By contrast, substituting sulfur atoms with oxygen atoms in 2H-MoS2 does not produce any significant change in the electronic band gap. We found that α-MoO3 with high sulfur concentrations (greater than 33%) is thermodynamically stabilized by H2S sulfidation and exhibit improved dielectric function, optical and charge transport properties and exciton binding energy for with respect to pristine α-MoO3. The carrier mobility is enhanced by such high sulfur concentrations induced by the delocalization of S-electronic states at the top of the valence band. For the oxygen substituted 2H-MoS2 bulk material, the optoelectronic properties, charge mobilities and charge separation are either unaltered or even degraded with respect to 2H-MoS2.
 Please wait while we load your content...
Please wait while we load your content...

