
α-Bi2Sn2O7: a potential room temperature n-type oxide thermoelectric†

Abstract
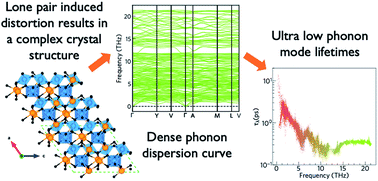
Interest in oxide thermoelectrics has been building due to their high thermal stability and earth-abundant constituent elements. However, the thermoelectric efficiency of flagship oxide materials remains comparatively low, and most materials only reach the maximum figure of merit, ZT, at very high temperatures, above those where the majority of low-grade industrial heat is emitted. It is important to identify thermoelectrics with high conversion efficiency closer to room temperature, particularly for lower-temperature applications such as in domestic heating, consumer electronics and electric vehicles. One of the main factors limiting the efficiency of oxide thermoelectrics is their large lattice thermal conductivities, which has inspired research into more structurally complex materials. In this study, we apply first-principles modelling to assess the low-temperature polymorph of Bi2Sn2O7 (α-Bi2Sn2O7) as a potential thermoelectric material, due to its complex crystal structure, which should suppress phonon transport, and the presence of Bi p and Sn s states in the conduction band, which should yield high electrical conductivity when donor (n) doped. Lattice-dynamics calculations using third-order perturbation theory predict an ultralow room-temperature lattice thermal conductivity of 0.4 W m−1 K−1, the lowest ever predicted for an oxide material, and suggest that nanostructuring to a grain size of 5 nm could further decrease this to 0.28 W m−1 K−1. The ultralow lattice thermal conductivity gives α-Bi2Sn2O7 a maximum ZT of 0.36 at 385 K (0.46 with nanostructuring), which is the highest low-temperature value predicted for an oxide thermoelectric. Most importantly, our analysis highlights the relationship between the structural complexity, the chemical nature of the cation, and the short phonon lifetimes, and thus provides guidelines for identifying other novel high-performance oxide thermoelectrics.
 Please wait while we load your content...
Please wait while we load your content...

