
Defect chemistry of disordered solid-state electrolyte Li10GeP2S12†
 *ab
Shriram
Santhanagopalan
b
and
*ab
Shriram
Santhanagopalan
b
and
Abstract
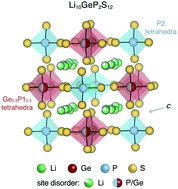
Several classes of materials, including thiophosphates, garnets, argyrodites, and anti-perovskites, have been considered as electrolytes for all-solid-state batteries. Native point defects and dopants play a critical role in impeding or facilitating fast ion conduction in these solid electrolytes. Despite its significance, comprehensive studies of the native defect chemistry of well-known solid electrolytes is currently lacking, in part due their compositional and structural complexity. Most of these solid-state electrolytes exhibit significant structural disorder, which requires careful consideration when modeling the point defect energetics. In this work, we model the native defect chemistry of a disordered solid electrolyte, Li10GeP2S12 (LGPS), by uniquely combining ensemble statistics, accurate electronic structure, and modern first-principles defect calculations. We find that VLi, Lii, and PGe are the dominant defects. From these calculations, we determine the statistics of defect energetics; formation energies of the dominant defects vary over ∼140 meV. Combined with previously reported ab initio molecular dynamics simulations, we find that anti-sites PGe promote Li ion conductivity, suggesting LGPS growth under P-rich/Ge-poor conditions will enhance ion conductivity. To this end, we offer practical experimental guides to enhance ion conductivity.
- This article is part of the themed collection: Editor’s Choice: Solid-state ion conductors
 Please wait while we load your content...
Please wait while we load your content...

