
Elucidating hydrogen storage properties of two-dimensional siligraphene (SiC8) monolayers upon selected metal decoration†

Abstract
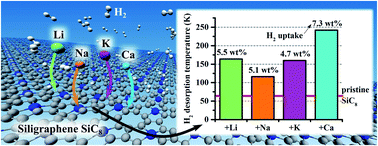
Density functional theory calculations with dispersion corrections were employed to investigate the hydrogen (H2) adsorptive properties of siligraphene (SiC8), pristine and decorated with selected alkali (Li, Na, and K) and alkaline-earth (Be, Mg, and Ca) metals. We found that all the considered metals (Me), except Mg and Be, bind strongly to SiC8 even at high doping concentrations (SiC8Me2) by donating a major portion of their valence electrons to SiC8. Ab initio molecular dynamics (AIMD) simulations confirmed the thermal stabilities of SiC8Me2 (Me = Li, Na, K, Ca) at 300 K. We showed that Li, Na, and Ca-doped SiC8 adsorbed multiple H2 molecules with binding energies (Ebind) at least two times stronger than that of the pristine SiC8 (Epristinebind = −70 meV per H2). Overall, both SiC8Li2 and SiC8Ca2 adsorbed two and four H2 molecules per metal adatom, respectively, having Ebind within the desirable range for an effective adsorption/desorption process. The resulting gravimetric densities of SiC8Li2 and SiC8Ca2 were 5.5 wt% and 7.3 wt%, respectively, surpassing the U.S. Department of Energy's 2025 goal of 5.5 wt%. The estimated H2 desorption temperatures exceed substantially the boiling point of liquid nitrogen, confirming the potential of light metal decorated SiC8 as a promising material for H2 storage.
 Please wait while we load your content...
Please wait while we load your content...

