
Unveiling the molecular mechanism of SARS-CoV-2 main protease inhibition from 137 crystal structures using algebraic topology and deep learning†
 *bcd
*bcd
Abstract
Currently, there is neither effective antiviral drugs nor vaccine for coronavirus disease 2019 (COVID-19) caused by acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Due to its high conservativeness and low similarity with human genes, SARS-CoV-2 main protease (Mpro) is one of the most favorable drug targets. However, the current understanding of the molecular mechanism of Mpro inhibition is limited by the lack of reliable binding affinity ranking and prediction of existing structures of Mpro–inhibitor complexes. This work integrates mathematics (i.e., algebraic topology) and deep learning (MathDL) to provide a reliable ranking of the binding affinities of 137 SARS-CoV-2 Mpro inhibitor structures. We reveal that Gly143 residue in Mpro is the most attractive site to form hydrogen bonds, followed by Glu166, Cys145, and His163. We also identify 71 targeted covalent bonding inhibitors. MathDL was validated on the PDBbind v2016 core set benchmark and a carefully curated SARS-CoV-2 inhibitor dataset to ensure the reliability of the present binding affinity prediction. The present binding affinity ranking, interaction analysis, and fragment decomposition offer a foundation for future drug discovery efforts.
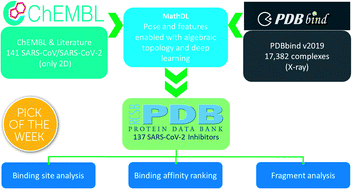
- This article is part of the themed collections: Coronavirus articles - free to access collection, 2020 ChemSci Pick of the Week Collection and 2020 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

