
Polymorph exploration of bismuth stannate using first-principles phonon mode mapping†

Abstract
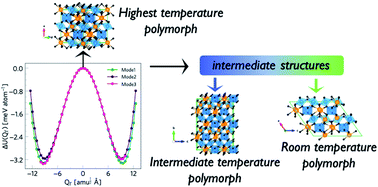
Accurately modelling polymorphism in crystalline solids remains a key challenge in computational chemistry. In this work, we apply a theoretically-rigorous phonon mode-mapping approach to understand the polymorphism in the ternary metal oxide Bi2Sn2O7. Starting from the high-temperature cubic pyrochlore aristotype, we systematically explore the structural potential-energy surface and recover the two known low-temperature phases alongside three new metastable phases, together with the transition pathways connecting them. This first-principles lattice-dynamics method is completely general and provides a practical means to identify and characterise the stable polymorphs and phase transitions in materials with complex crystal structures.
 Please wait while we load your content...
Please wait while we load your content...

