
Structure and solvation of confined water and water–ethanol clusters within microporous Brønsted acids and their effects on ethanol dehydration catalysis†

Abstract
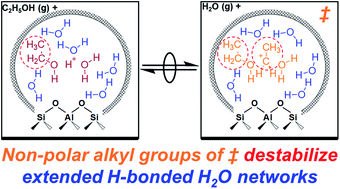
Aqueous-phase reactions within microporous Brønsted acids occur at active centers comprised of water-reactant-clustered hydronium ions, solvated within extended hydrogen-bonded water networks that tend to stabilize reactive intermediates and transition states differently. The effects of these diverse clustered and networked structures were disentangled here by measuring turnover rates of gas-phase ethanol dehydration to diethyl ether (DEE) on H-form zeolites as water pressure was increased to the point of intrapore condensation, causing protons to become solvated in larger clusters that subsequently become solvated by extended hydrogen-bonded water networks, according to in situ IR spectra. Measured first-order rate constants in ethanol quantify the stability of SN2 transition states that eliminate DEE relative to (C2H5OH)(H+)(H2O)n clusters of increasing molecularity, whose structures were respectively determined using metadynamics and ab initio molecular dynamics simulations. At low water pressures (2–10 kPa H2O), rate inhibition by water (−1 reaction order) reflects the need to displace one water by ethanol in the cluster en route to the DEE-formation transition state, which resides at the periphery of water–ethanol clusters. At higher water pressures (10–75 kPa H2O), water–ethanol clusters reach their maximum stable size ((C2H5OH)(H+)(H2O)4–5), and water begins to form extended hydrogen-bonded networks; concomitantly, rate inhibition by water (up to −3 reaction order) becomes stronger than expected from the molecularity of the reaction, reflecting the more extensive disruption of hydrogen bonds at DEE-formation transition states that contain an additional solvated non-polar ethyl group compared to the relevant reactant cluster, as described by non-ideal thermodynamic formalisms of reaction rates. Microporous voids of different hydrophilic binding site density (Beta; varying H+ and Si–OH density) and different size and shape (Beta, MFI, TON, CHA, AEI, FAU), influence the relative extents to which intermediates and transition states disrupt their confined water networks, which manifest as different kinetic orders of inhibition at high water pressures. The confinement of water within sub-nanometer spaces influences the structures and dynamics of the complexes and extended networks formed, and in turn their ability to accommodate the evolution in polarity and hydrogen-bonding capacity as reactive intermediates become transition states in Brønsted acid-catalyzed reactions.
 Please wait while we load your content...
Please wait while we load your content...

