
Interpretable molecular models for molybdenum disulfide and insight into selective peptide recognition†

Abstract
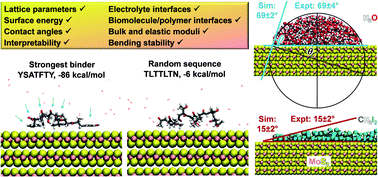
Molybdenum disulfide (MoS2) is a layered material with outstanding electrical and optical properties. Numerous studies evaluate the performance in sensors, catalysts, batteries, and composites that can benefit from guidance by simulations in all-atom resolution. However, molecular simulations remain difficult due to lack of reliable models. We introduce an interpretable force field for MoS2 with record performance that reproduces structural, interfacial, and mechanical properties in 0.1% to 5% agreement with experiments. The model overcomes structural instability, deviations in interfacial and mechanical properties by several 100%, and empirical fitting protocols in earlier models. It is compatible with several force fields for molecular dynamics simulation, including the interface force field (IFF), CVFF, DREIDING, PCFF, COMPASS, CHARMM, AMBER, and OPLS-AA. The parameters capture polar covalent bonding, X-ray structure, cleavage energy, infrared spectra, bending stability, bulk modulus, Young's modulus, and contact angles with polar and nonpolar solvents. We utilized the models to uncover the binding mechanism of peptides to the MoS2 basal plane. The binding strength of several 7mer and 8mer peptides scales linearly with surface contact and replacement of surface-bound water molecules, and is tunable in a wide range from −86 to −6 kcal mol−1. The binding selectivity is multifactorial, including major contributions by van-der-Waals coordination and charge matching of certain side groups, orientation of hydrophilic side chains towards water, and conformation flexibility. We explain the relative attraction and role of the 20 amino acids using computational and experimental data. The force field can be used to screen and interpret the assembly of MoS2-based nanomaterials and electrolyte interfaces up to a billion atoms with high accuracy, including multiscale simulations from the quantum scale to the microscale.
 Please wait while we load your content...
Please wait while we load your content...

