
Simulating protein–ligand binding with neural network potentials†
 a
and
Christopher N.
Rowley
*a
a
and
Christopher N.
Rowley
*a
Abstract
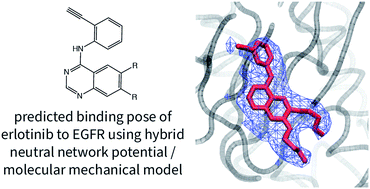
Drug molecules adopt a range of conformations both in solution and in their protein-bound state. The strain and reduced flexibility of bound drugs can partially counter the intermolecular interactions that drive protein–ligand binding. To make accurate computational predictions of drug binding affinities, computational chemists have attempted to develop efficient empirical models of these interactions, although these methods are not always reliable. Machine learning has allowed the development of highly-accurate neural-network potentials (NNPs), which are capable of predicting the stability of molecular conformations with accuracy comparable to state-of-the-art quantum chemical calculations but at a billionth of the computational cost. Here, we demonstrate that these methods can be used to represent the intramolecular forces of protein-bound drugs within molecular dynamics simulations. These simulations are shown to be capable of predicting the protein–ligand binding pose and conformational component of the absolute Gibbs energy of binding for a set of drug molecules. Notably, the conformational energy for anti-cancer drug erlotinib binding to its target was found to be considerably overestimated by a molecular mechanical model, while the NNP predicts a more moderate value. Although the ANI-1ccX NNP was not trained to describe ionic molecules, reasonable binding poses are predicted for charged ligands, but this method is not suitable for modeling charged ligands in solution.
- This article is part of the themed collection: 2020 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

