
The role of an interface in stabilizing reaction intermediates for hydrogen evolution in aprotic electrolytes†

Abstract
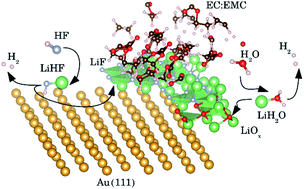
By combining idealized experiments with realistic quantum mechanical simulations of an interface, we investigate electro-reduction reactions of HF, water and methanesulfonic acid (MSA) on the single crystal (111) facets of Au, Pt, Ir and Cu in organic aprotic electrolytes, 1 M LiPF6 in EC/EMC 3:7W (LP57), the aprotic electrolyte commonly used in Li-ion batteries, 1 M LiClO4 in EC/EMC 3:7W and 0.2 M TBAPF6 in 3 : 7 EC/EMC. In our previous work, we have established that LiF formation, accompanied by H2 evolution, is caused by a reduction of HF impurities and requires the presence of Li at the interface, which catalyzes the HF dissociation. In the present paper, we find that the measured potential of the electrochemical response for these reduction reactions correlates with the work function of the electrode surfaces and that the work function determines the potential for Li+ adsorption. The reaction path is investigated further by electrochemical simulations suggesting that the overpotential of the reaction is related to stabilizing the active structure of the interface having adsorbed Li+. Li+ is needed to facilitate the dissociation of HF which is the source of protons. Further experiments on other proton sources, water and methanesulfonic acid, show that if the hydrogen evolution involves negatively charged intermediates, F− or HO−, a cation at the interface can stabilize them and facilitate the reaction kinetics. When the proton source is already significantly dissociated (in the case of a strong acid), there is no negatively charged intermediate and thus the hydrogen evolution can proceed at much lower overpotentials. This reveals a situation where the overpotential for electrocatalysis is related to stabilizing the active structure of the interface, facilitating the reaction rather than providing the reaction energy.
 Please wait while we load your content...
Please wait while we load your content...

