
Free energy of proton transfer at the water–TiO2 interface from ab initio deep potential molecular dynamics†

Abstract
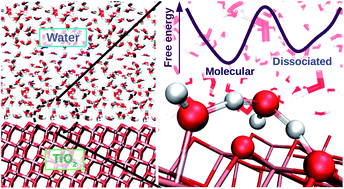
TiO2 is a widely used photocatalyst in science and technology and its interface with water is important in fields ranging from geochemistry to biomedicine. Yet, it is still unclear whether water adsorbs in molecular or dissociated form on TiO2 even for the case of well-defined crystalline surfaces. To address this issue, we simulated the TiO2–water interface using molecular dynamics with an ab initio-based deep neural network potential. Our simulations show a dynamical equilibrium of molecular and dissociative adsorption of water on TiO2. Water dissociates through a solvent-assisted concerted proton transfer to form a pair of short-lived hydroxyl groups on the TiO2 surface. Molecular adsorption of water is ΔF = 8.0 ± 0.9 kJ mol−1 lower in free energy than the dissociative adsorption, giving rise to a 5.6 ± 0.5% equilibrium water dissociation fraction at room temperature. Due to the relevance of surface hydroxyl groups to the surface chemistry of TiO2, our model might be key to understanding phenomena ranging from surface functionalization to photocatalytic mechanisms.
- This article is part of the themed collections: Most popular 2019-2020 catalysis articles and 2020 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

