
Excited-state proton-coupled electron transfer within ion pairs†

Abstract
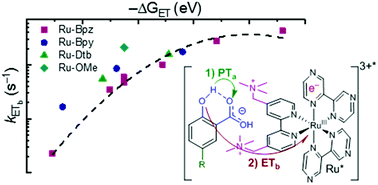
The use of light to drive proton-coupled electron transfer (PCET) reactions has received growing interest, with recent focus on the direct use of excited states in PCET reactions (ES-PCET). Electrostatic ion pairs provide a scaffold to reduce reaction orders and have facilitated many discoveries in electron-transfer chemistry. Their use, however, has not translated to PCET. Herein, we show that ion pairs, formed solely through electrostatic interactions, provide a general, facile means to study an ES-PCET mechanism. These ion pairs formed readily between salicylate anions and tetracationic ruthenium complexes in acetonitrile solution. Upon light excitation, quenching of the ruthenium excited state occurred through ES-PCET oxidation of salicylate within the ion pair. Transient absorption spectroscopy identified the reduced ruthenium complex and oxidized salicylate radical as the primary photoproducts of this reaction. The reduced reaction order due to ion pairing allowed the first-order PCET rate constants to be directly measured through nanosecond photoluminescence spectroscopy. These PCET rate constants saturated at larger driving forces consistent with approaching the Marcus barrierless region. Surprisingly, a proton-transfer tautomer of salicylate, with the proton localized on the carboxylate functional group, was present in acetonitrile. A pre-equilibrium model based on this tautomerization provided non-adiabatic electron-transfer rate constants that were well described by Marcus theory. Electrostatic ion pairs were critical to our ability to investigate this PCET mechanism without the need to covalently link the donor and acceptor or introduce specific hydrogen bonding sites that could compete in alternate PCET pathways.
- This article is part of the themed collection: Papers celebrating the birthday and achievements of Professor Michael Wasielewski
 Please wait while we load your content...
Please wait while we load your content...

