
The hunt for reactive alkynes in bio-orthogonal click reactions: insights from mechanochemical and conceptual DFT calculations†

Abstract
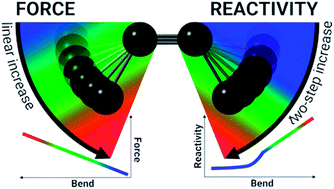
In our effort to implement the mechanical force used to activate single molecules in mechanochemistry in the context of conceptual density functional theory, we present a theoretical investigation of strained alkynes for rationalizing structural trends as well as the reactivity of cyclic alkynes that are of great importance in in vivo click reactions. The strain on the triple bond in cyclic alkynes is modeled by angular constraints in a 2-butyne fragment and the corresponding bending force is calculated by means of an extended COGEF (constrained geometries simulate external forces) model. In general, the force required to bend the triple bond is smaller with electron-withdrawing groups on the propargylic C-atom, which elegantly results in smaller angles around the triple bond in cyclic alkynes with such substitution pattern. By means of conceptual DFT descriptors, the electrophilic and nucleophilic character of bent triple bonds was investigated revealing moderate activation for small distortions from the linear geometry (0° to 15°) and a drastically more reactive π-space if the triple bond is bent further. This analysis of the intrinsic reactivity of the triple bond is in line with experimental observations, explaining the reactive nature of cyclooctynes and cycloheptynes, whereas larger cyclic systems do not drastically activate the triple bond.
- This article is part of the themed collection: Bioorthogonal and click chemistry: Celebrating the 2022 Nobel Prize in Chemistry
 Please wait while we load your content...
Please wait while we load your content...

