
Predicting adsorption selectivities from pure gas isotherms for gas mixtures in metal–organic frameworks†

Abstract
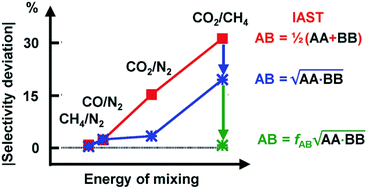
We perform Grand Canonical Monte Carlo simulations on a lattice of Mg2+ sites (GCMC) for adsorption of four binary A/B mixtures, CH4/N2, CO/N2, CO2/N2, and CO2/CH4, in the metal–organic framework Mg2(2,5-dioxidobenzedicarboxylate), also known as CPO-27–Mg or Mg–MOF-74. We present a mean field co-adsorption isotherm model and show that its predictions agree with the GCMC results if the same quantum chemical ab initio data are used for Gibbs free energies of adsorption at the individual sites and for lateral interaction energies between the same, A⋯A and B⋯B, and unlike, A⋯B, adsorbed molecules. We use both approaches to test the assumption underlying Ideal Adsorbed Solution Theory (IAST), namely approximating A⋯B interaction energies as the arithmetic mean of A⋯A and B⋯B interaction energies. While IAST works well for mixtures with weak lateral interactions, CH4/N2 and CO/N2, the deviations are large for mixtures with stronger lateral interactions, CO2/N2 and CO2/CH4. Motivated by the theory of London dispersion forces, we propose use of the geometric mean instead of the arithmetic mean and achieve substantial improvements. For CO2/CH4, the lateral interactions become anisotropic. To include this in the geometric mean co-adsorption model, we introduce an anisotropy factor. We propose a protocol, named co-adsorption mean field theory (CAMT), for co-adsorption selectivity prediction from known (experiment or simulation) pure component isotherms which is similar to the IAST protocol but uses the geometric mean to approximate mixed pair interaction energies and yields improved results for non-ideal mixtures.
- This article is part of the themed collection: Editor’s Choice – Jinlong Gong
 Please wait while we load your content...
Please wait while we load your content...

