
Drug–target affinity prediction using graph neural network and contact maps
 *a
*a
Abstract
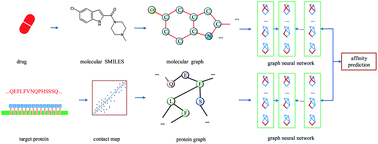
Computer-aided drug design uses high-performance computers to simulate the tasks in drug design, which is a promising research area. Drug–target affinity (DTA) prediction is the most important step of computer-aided drug design, which could speed up drug development and reduce resource consumption. With the development of deep learning, the introduction of deep learning to DTA prediction and improving the accuracy have become a focus of research. In this paper, utilizing the structural information of molecules and proteins, two graphs of drug molecules and proteins are built up respectively. Graph neural networks are introduced to obtain their representations, and a method called DGraphDTA is proposed for DTA prediction. Specifically, the protein graph is constructed based on the contact map output from the prediction method, which could predict the structural characteristics of the protein according to its sequence. It can be seen from the test of various metrics on benchmark datasets that the method proposed in this paper has strong robustness and generalizability.
 Please wait while we load your content...
Please wait while we load your content...

