
Electronic, magnetic and optical properties of MnPX3 (X = S, Se) monolayers with and without chalcogen defects: a first-principles study†
 *a
and
Elena
Voloshina
*ac
*a
and
Elena
Voloshina
*ac
Abstract
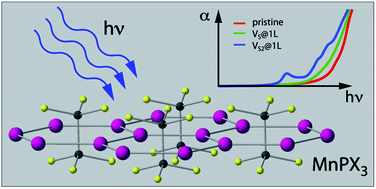
Based on density functional theory (DFT), we performed first-principles studies on the electronic structure, magnetic state and optical properties of two-dimensional (2D) transition-metal phosphorous trichalcogenides MnPX3 (X = S and Se). The calculated interlayer cleavage energies of the MnPX3 monolayers indicate the energetic possibility to be exfoliated from the bulk phase, with good dynamical stability confirmed by the absence of imaginary contributions in the phonon spectra. The MnPX3 monolayers are both Néel antiferromagnetic (AFM) semiconductors with direct band gaps falling into the visible optical spectrum. Magnetic interaction parameters were extracted within the Heisenberg model to investigate the origin of the AFM state. Three in-plane magnetic exchange parameters play an important role in the robust AFM configuration of Mn ions. The Néel temperatures (TN) were estimated by means of Monte Carlo simulations, obtaining theoretical TN values of 103 K and 80 K for 2D MnPS3 and MnPSe3, respectively. With high spin state Mn ions arranged in honeycomb lattices, the spin-degenerated band structures exhibit valley polarisation and were investigated in different biaxial in-plain strains, considering the spin-orbital coupling (SOC). 2D MnPX3 monolayers show excellent performance in terms of the optical properties, and the absorption spectra were discussed in detail to find the transition mechanism. Different amounts and configurations of chalcogen vacancies were introduced into the MnPX3 monolayers, and it was found that the electronic structures are heavily affected depending on the vacancy geometric structure, leading to different magnetic state and absorption spectra of defected MnPX3 systems.
 Please wait while we load your content...
Please wait while we load your content...

