
Mechanism and origins of stereo- and enantioselectivities of palladium-catalyzed hydroamination of racemic internal allenes via dynamic kinetic resolution: a computational study†

Abstract
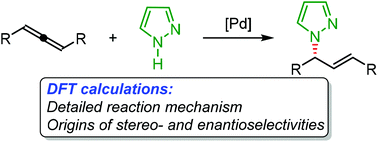
Density functional theory calculations have been performed to investigate the Pd-catalyzed hydroamination of racemic internal allenes with pyrazoles. The computations show that the originally proposed reaction mechanisms initiated by the formation of the Pd(II)-hydride complexes are kinetically infeasible. Instead, it was suggested that the Pd(0) intermediate, generated by C–Cl bond formation via the outer-sphere nucleophilic attack, is the active catalyst of the reaction. The protonation of the central carbon of the Pd(0)-coordinated allene using pyrazole as the proton source followed by C–N bond formation through the outer-sphere nucleophilic attack of the pyrazole ligand on the η3-allyl ligand could afford the final hydroamination products. The outer-sphere nucleophilic attack constitutes the stereo- and enantioselectivity-determining step of the overall reaction. The experimentally observed stereo- and enantioselectivities were reproduced quite well by the calculations, which were found to be attributed to the steric and electronic effects. The steric repulsion between the phenyl group of the (R)-SEGPHOS ligand and the methylene group of the allene moiety, together with the additional C–H⋯π and C–H⋯N interactions, results in the (E)–(S)-transition state being more favorable than the other transition states, leading to the experimental outcomes.
 Please wait while we load your content...
Please wait while we load your content...

