
Dual reactivity of B(C6F5)3 enables the silylative cascade conversion of N-aryl piperidines to sila-N-heterocycles: DFT calculations†
 *bc
and
Li
Dang
*a
*bc
and
Li
Dang
*a
Abstract
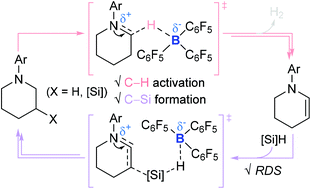
The first catalytic access to bridged sila-N-heterocycles from piperidines via cascade sp3 and sp2 C–Si bond formation all mediated by a single catalyst B(C6F5)3 (A) has been recently developed by Park and Chang. Described herein is a theoretical study of the B(C6F5)3-catalyzed silylative cascade conversion of N-aryl piperidines (H) to afford polycyclic azasilaheterocycles with a strong emphasis on the dual reactivity of B(C6F5)3. The DFT calculations show that the catalyzed reaction involves three steps of the cascade reaction: (i) the formation of tetrahydropyridine (I) upon dehydrogenation of the piperidine; (ii) β-selective hydrosilylation of tetrahydropyridine; (iii) an intramolecular sila-Friedel–Crafts reaction to form a bridged sp2 C–Si bond. The N-silyl piperidinium borohydride (A′) turns out to be the thermodynamically favored, resting species of the overall reaction, which is consistent with the observation of a species such as A′ during borane catalysis. The DFT calculations suggest that the β-selective hydrosilylation of I is the rate-determining step in the entire catalytic cycle, while the liberated I reacts with B(C6F5)3 to form a zwitterion consisting of iminium and borate units (O). The calculated sila-Friedel–Crafts reactions of a range of presupposed silylated (cyclic)aza intermediates imply the importance of structural integrity on the piperidinyl unit of substrates.
 Please wait while we load your content...
Please wait while we load your content...

