
Rhodium-catalyzed ene-cycloisomerization of allylic-sulfide-tethered alkylidenecyclopropanes: DFT analysis of origins of regio- and diastereo-selectivities†
 *a
*a
Abstract
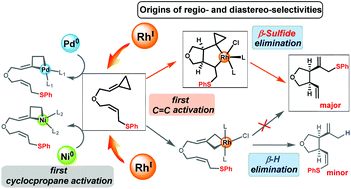
Rhodium-catalyzed ene-cycloisomerization of allylic-sulfide-tethered alkylidenecyclopropanes (ACPs) is an efficient method for late-transition-metal-mediated β-sulfide elimination. The density functional theory (DFT) method was used to investigate the mechanism, and regio- and diastereo-selectivities of this type of reaction. The computational results showed that the unique control of the regio- and diastereo-selectivities of this reaction can be attributed to an unconventional reaction mechanism. Instead of the commonly accepted mechanism, which involves initial ring opening of the ACP, carbometallation, β-sulfide elimination, and thioether migration, the Rh(I)-catalyzed ene-cycloisomerization reaction occurs via activation of the ACP double bond, β-sulfide elimination, and the simultaneous thioether transfer and ring opening of the cyclopropyl group. Importantly, the calculation results explain why initial ACP double-bond activation was achieved with a Rh(I) catalyst but not with Pd(0) and Ni(0) catalysts. This mechanism does not occur with Pd(0) and Ni(0) catalysts because of steric effects.
 Please wait while we load your content...
Please wait while we load your content...

