
C–F bond arylation of fluoroarenes catalyzed by Pd0 phosphine complexes: theoretical insight into regioselectivity, reactivity, and prediction of ligands†

Abstract
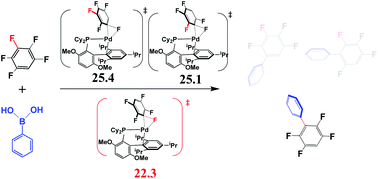
Palladium-catalyzed C–F bond arylation of pentafluorobenzene was theoretically investigated as an example of aryl–F bond functionalization. DFT computations show that C3-regioselective arylation of pentafluorobenzene occurs more favorably than C1 and C2-ones as reported experimentally, through oxidative addition of the C–F bond to Pd0 species, transmetalation and reductive elimination of the C–C bond. Oxidative addition of the C–F bond is the rate-determining and regioselectivity-determining step. The lower energy transition state of the oxidative addition of the C3–F bond (TS-C3) arises from a larger stabilization energy between Pd0(BrettPhos) and distorted pentafluorobenzene moieties in TS-C3 than those in TS-C1 and TS-C2. The larger stabilization energy is a result of a lower σ* orbital energy of the distorted C3–F bond than those of C1–F and C2–F bonds, which leads to a larger charge transfer from the Pd dπ orbital to the σ* orbital of the C3–F bond. The results suggest that both σ* orbital energy and bond dissociation energy are important factors for determining the reactivity of the C–F bond. Also, the activation barriers of the C–F bond with different substitution groups follow the order: NO2 < COOMe < CN ∼ CF3 < F, which is approximately consistent with the order of electron-withdrawing ability of these groups. It is theoretically predicted here that NMe2-substituted BrettPhos is better for C–F bond cleavage than BrettPhos, where three NMe2 groups are introduced to BrettPhos instead of the isopropyl groups.
 Please wait while we load your content...
Please wait while we load your content...

