
Electronic, vibrational, and charge-transport properties of benzothienobenzothiophene–TCNQ co-crystals†‡
 a
Caitlin
Hanson,
§a
Nathan
Corbin,
¶a
Jean-Luc
Brédas
*ab
and
Veaceslav
Coropceanu
*ab
a
Caitlin
Hanson,
§a
Nathan
Corbin,
¶a
Jean-Luc
Brédas
*ab
and
Veaceslav
Coropceanu
*ab
Abstract
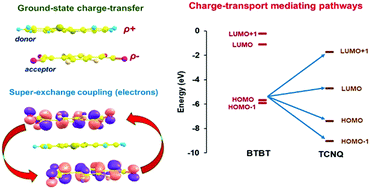
The electronic, vibrational, and charge-transport properties of a series of benzothieno-benzothiophene (BTBT)–FmTCNQ (m = 0, 2, 4) and diCnBTBT–FmTCNQ (n = 8, 12; m = 0, 4) donor–acceptor (DA) co-crystals have been investigated by means of density functional theory calculations. The electronic-structure calculations predict wide conduction bands and small effective masses for electrons along the DA stacking directions. The results indicate that the increase in the number of F atoms on the acceptor molecules results in an increase of superexchange couplings along the DA stacks, while the addition of the alkyl side chains results in a decrease of through-space transfer integrals between neighboring stacks. Time-dependent density functional theory calculations of the optical properties describe the lowest two optical transitions as having a charge-transfer character and being related to the two electronic coupling pathways that contribute to the superexchange couplings. The results also indicate that the ionicity parameter in the diCnBTBT–FmTCNQ cocrystals is somewhat larger than in the BTBT analogues. Overall, we find that DFT calculations based on periodic boundary conditions are a reliable tool to estimate the ionicity parameter in DA cocrystals.
- This article is part of the themed collections: Celebrating our 2021 Prizewinners and Celebrating Prof. Fred Wudl’s 80th Birthday
 Please wait while we load your content...
Please wait while we load your content...

