
Thermodynamic insights into interfacial interactions in TiN/amorphous Al2O3 heterostructures: ab initio molecular dynamics and first principles investigation†
 *ad
*ad
Abstract
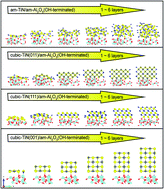
The nature and the mechanism of the film interaction with the substrate at the film/substrate interface are still far from being fully understood. Here, we demonstrate an ab initio investigation of the thermodynamic driving force across the initial phase among amorphous and crystalline structures in the film formation of TiN on an alumina substrate. Combining ab initio molecular dynamics (AIMD) and density functional theory (DFT), it provides evidence that the amorphous TiN indeed becomes thermodynamically stable at finite length scales (0.6 nm) as the conductive and adhesive layer on am-Al2O3. In other words, cubic TiN growth is thermodynamically restricted until the thickness reaches above 0.6 nm, and therefore the amorphous TiN film can maintain its conformal coverage under such conditions. The approach presented here can be used for computational design and discovery of new corrosion-resistant alloys and semiconductors, by providing a first-principles framework to search for ways to induce a stable conductive and adhesive TiN layer. Other possible applications are predicted to be in the area of thin-film deposition, especially, searching for predictive pathways for the selection of polymorphs during the deposition process.
 Please wait while we load your content...
Please wait while we load your content...

