
An ab initio approach on the asymmetric stacking of GaAs 〈111〉 nanowires grown by a vapor–solid method†

Abstract
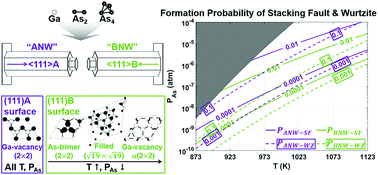
This study provides an ab initio thermodynamics approach to take a step forward in the theoretical modeling on the growth of GaAs nanowires. In order to understand the effects of growth conditions on the involvement of stacking faults and polytypism, we investigated the vapor-phase growth kinetics under arbitrary temperature–pressure conditions by combining the atomic-scale calculation with the thermodynamic treatment of a vapor–solid system. Considering entropy contribution and electronic energy, the chemical potential and surface energies of various reconstructions were calculated as a function of temperature and pressure, leading to the prediction of the change in Gibbs free energy at each stage of nucleation and growth. This enabled us to predict the temperature–pressure-dependent variation in nucleation rate and formation probability of possible stacking sequences: zinc blende, stacking faults, twin, and wurtzite. As a result, the formation probabilities of stacking faults and polytypism were found to decrease with increasing temperature or decreasing pressure, which agreed well with available experiments. In addition, by showing that the formation probability of the stacking defects in GaAs nanowires grown along the 〈111〉B direction is about ten times higher than that along the 〈111〉A direction, the intriguing asymmetric stacking behavior during the growth along the polar direction and its dependence on growth conditions were fundamentally elucidated. The proposed ab initio approach bridges the gap between atomic-scale static calculation at zero-temperature and kinetic growth process under arbitrary vapor-phase conditions, and thus will contribute to the nanoscale growth not only for GaAs nanowires but also for other materials.
 Please wait while we load your content...
Please wait while we load your content...

