
Chemical vapour deposition of graphene on copper–nickel alloys: the simulation of a thermodynamic and kinetic approach†

Abstract
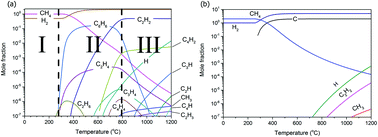
Chemical vapour deposition (CVD) of graphene on transition metals is generally believed to be the fabrication route best suited for the production of high-quality large-area graphene sheets. The mechanism of CVD graphene growth is governed by interactions in both the gas phase and at the surface. Here we present a simulation of the CVD graphene growth mechanism which includes thermodynamics, gas phase kinetics and the surface reaction in a sequential manner. The thermodynamic simulation shows that the deposition driving force is the greatest for high carbon to hydrogen ratios and reaches a maximum at around 850 °C. No graphene growth is observed below this temperature. The surface kinetic model also shows that below this temperature, the carbon surface concentration is less than the solubility limit, thus no film can grow. The effect of the reaction chamber geometry on the product concentrations was clear from the gas phase decomposition reactions. The gas residence times studied here (around 0.07 s) show that the optimum gas phase composition is far from that expected at thermodynamic equilibrium. The surface kinetics of CH4 reactions on Ni, Cu and Cu–Ni surfaces shows good agreement with the experimental results for different growth pressures (0.1 to 0.7 mbar), temperatures (600 to 1200 °C) and different Ni thicknesses (25–500 μm). Also, the model works well when substrates with various C solubilities are used. The thermodynamic and kinetic models described here can be used for the design of improved reactors to optimise the production of graphene with differing qualities, either single or multi-layer and sizes. More importantly, the transfer to a continuous process with a moving substrate should also be possible using the model if it is extended from 2D to 3D.
 Please wait while we load your content...
Please wait while we load your content...

