
An accurate free energy estimator: based on MM/PBSA combined with interaction entropy for protein–ligand binding affinity†

Abstract
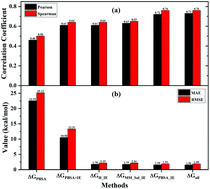
The molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method is constantly used to calculate the binding free energy of protein–ligand complexes, and has been shown to effectively balance computational cost against accuracy. The relative binding affinities obtained by the MM/PBSA approach are acceptable, while it usually overestimates the absolute binding free energy. This paper proposes four free energy estimators based on the MM/PBSA for enthalpy change combined with interaction entropy (IE) for entropy change using different weights for individual energy terms. The ΔGPBSA_IE method is determined to be an optimal estimator based on its performance in terms of the correlation between experimental and theoretical values and error estimations. This approach is optimized using high-quality experimental values from a training set containing 84 protein–ligand systems, and the coefficients for the sum of electrostatic energy and polar solvation free energy, van der Waals (vdW) energy, non-polar solvation energy and entropy change are obtained by multivariate linear fitting to the corresponding experimental values. A comparison between the traditional MM/PBSA method and this method shows that the correlation coefficient is improved from 0.46 to 0.72 and the slope of the regression line increases from 0.10 to 1.00. More importantly, the mean absolute error (MAE) is significantly reduced from 22.52 to 1.59 kcal mol−1. Furthermore, the numerical stability of this method is validated on a test set with a similar correlation coefficient, slope and MAE to those of the training set. Based on the above advantages, the ΔGPBSA_IE method can be a powerful tool for a reliable and accurate estimation of binding free energy and plays a significant role in a detailed energetic investigation of protein–ligand interaction.
- This article is part of the themed collection: Editor’s Choice: Computational studies of nanomaterials for energy, catalysis and electronics
 Please wait while we load your content...
Please wait while we load your content...

