
The chemical effect goes resonant – a full quantum mechanical approach on TERS†
 a
Mostafa
Abasifard,
a
Volker
Deckert,
abc
Stefanie
Gräfe
*a
and
Stephan
Kupfer
*a
a
Mostafa
Abasifard,
a
Volker
Deckert,
abc
Stefanie
Gräfe
*a
and
Stephan
Kupfer
*a
Abstract
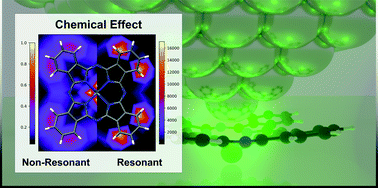
Lately, experimental evidence of unexpectedly extremely high spatial resolution of tip-enhanced Raman scattering (TERS) has been demonstrated. Theoretically, two different contributions are discussed: an electromagnetic effect, leading to a spatially confined near field due to plasmonic excitations; and the so-called chemical effect originating from the locally modified electronic structure of the molecule due to the close proximity of the plasmonic system. Most of the theoretical efforts have concentrated on the electromagnetic contribution or the chemical effect in case of non-resonant excitation. In this work, we present a fully quantum mechanical description including non-resonant and resonant chemical contributions as well as charge-transfer phenomena of these molecular-plasmonic hybrid systems at the density functional and the time-dependent density functional level of theory. We consider a surface-immobilized tin(II) phthalocyanine molecule as the molecular system, which is minutely scanned by a plasmonic tip, modeled by a single silver atom. These different relative positions of the Ag atom to the molecule lead to pronounced alterations of the Raman spectra. These Raman spectra vary substantially, both in peak positions and several orders of magnitude in the intensity patterns under non-resonant and resonant conditions, and also, depending on, which electronic states are addressed. Our computational approach reveals that unique – non-resonant and resonant – chemical interactions among the tip and the molecule significantly alter the TERS spectra and are mainly responsible for the high, possibly sub-Angstrom spatial resolution.
 Please wait while we load your content...
Please wait while we load your content...

