
Modeling generation and growth of iron oxide nanoparticles from representative precursors through ReaxFF molecular dynamics†
 *a
and
Susanna
Monti
b
*a
and
Susanna
Monti
b
Abstract
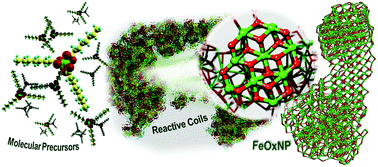
Detailed dynamical characterization of the mechanisms responsible for the formation and growth of iron oxide nanoparticles remains a significant challenge not only for experimental techniques but also for theoretical methodologies due to the nanoparticle size, long simulation times, and complexity of the environments. In this work, we have designed a fast computational protocol based on atomistic reactive molecular dynamics, which is capable of simulating the whole synthetic and proliferation process of the nanoparticles (greater than 10 nm) in a homogeneous medium from organometallic precursors. We have defined appropriate growth accelerating strategies based on the observed reactions, which consisted of the formation of Fe–O–Fe bridges, linking separate precursors, and Fe˙ and FeO˙ radicals. This reduced drastically the computational time allowing the simulation of NPs made of thousands of atoms (full nanometric range). We have identified the most probable reaction environments and summarized them under two distinct conditions: reductive and oxidative. The first one leads to the formation of nanoparticles with FeO stoichiometry typical of wustite, whereas the second one stabilizes stoichiometries between Fe3O4 (magnetite), and Fe2O3 (maghemite). In the latter case, the obtained NPs adopted, from the very early stages of the growth process, a cubic crystalline structure, typical of the oxidized FeOx bulk phases. The excellent agreement of our results with the experimental data demonstrates that the proposed protocol can provide a powerful predictive tool to describe structural features developed by the metal oxide nanoparticles and establish clear structure–property relationships.
 Please wait while we load your content...
Please wait while we load your content...

