
Global and local minima of protonated acetonitrile clusters†
 *ab
and
Jeanet
Conradie
a
*ab
and
Jeanet
Conradie
a
Abstract
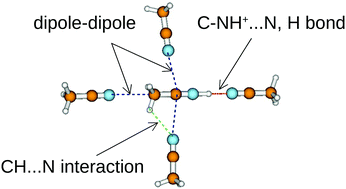
Understanding the proton transfer processes in acetonitrile is subjected to the knowledge of the structures of protonated acetonitrile clusters, H+(MeCN)n. Despite their importance, there is no clearly reported investigation of the structures of the protonated acetonitrile clusters, unlike the protonated water clusters or the protonated ammonia clusters. In this work, we thoroughly explored the potential energy surfaces (PESs) of the protonated acetonitrile clusters up to the decamer (n = 10) using three incremental levels of theory. We started by exploring the PESs of the protonated acetonitrile clusters using classical molecular dynamic to generate initial structures. Then, the generated structures have been fully optimized using a density functional theory (DFT) functional, MN15, associated to the 6-31++G(d,p) basis set. Finally, to gain more insights on the location of the global minima energy structures, all the structures of clusters up to the heptamer (n = 7) have been fully re-optimized at the MP2/aug-cc-pVDZ level of theory. As a result, we reported several local and global minima energy structures of the protonated acetonitrile clusters that have not been reported previously. The results show that the structures of the protonated acetonitrile clusters are stabilized by strong H+⋯N hydrogen bonds, anti-parallel dimers, dipole–dipole and CH⋯N interactions. Moreover, we noted that planar and branched isomers are the most favored for clusters lower than the hexamer, while larger-sized clusters favored compact isomers. To derive temperature effects on the stability of the investigated structures, we have reported the temperature-dependent isomer distributions of all the investigated clusters.
 Please wait while we load your content...
Please wait while we load your content...

