
Computationally exploring novel xanthine oxidase inhibitors using docking-based 3D-QSAR, molecular dynamics, and virtual screening†

Abstract
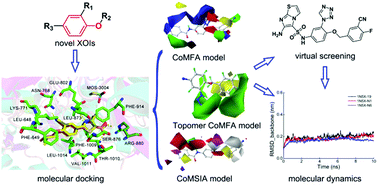
Several novel series of xanthine oxidase (XO) inhibitors (XOIs) with nitrogen-containing heterocycles have been reported recently. To better comprehend the three-dimensional quantitative structure–activity relationships (3D-QSAR) and mechanisms of action of these novel XOIs, in this study, a systematic modeling study was performed on 53 XOIs with diverse structures using the docking-based 3D-QSAR, virtual screening, and molecular dynamics (MD) methods. The docking results revealed that these XOIs could form hydrogen bonds, π–π stacking, and hydrophobic interactions at the binding site of XO, which might be important for maintaining their inhibitory activities. The 3D-QSAR models, comprising the comparative molecular field analysis (q2 = 0.741, R2 = 0.988, and rpred2 = 0.940), comparative molecular similarity indices analysis (q2 = 0.846, R2 = 0.982, and rpred2 = 0.916), and Topomer CoMFA (q2 = 0.947, R2 = 0.983, and rpred2 = 0.909) models, exhibited satisfying quality and excellent predictive ability. Six XOI hit compounds (N1–N6) with higher docking-scores and better predicted activities were obtained by virtual screening. The MD results indicated that compound N6 could stably bind with the XO and might be a promising XOI candidate. This study could provide useful information and theoretical guidance for the design and development of novel XOIs.
 Please wait while we load your content...
Please wait while we load your content...

