
DFT and hybrid-DFT calculations on the electronic properties of vanadate materials: theory meets experiments†

Abstract
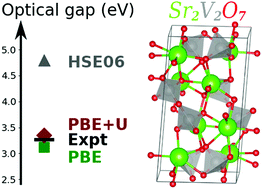
Herein is presented a theoretical study of the electronic structure and optical properties of six vanadium oxides: Sr2V2O7, Ba2V2O7, Ca2VO4Cl, Sr2VO4Cl, Mg3V2O8 and Zn3V2O8. Simulations were performed within the Density Functional Theory (DFT) framework. Properties were computed using the Generalized Gradient Approximation (GGA) PBE functional, a Hubbard-like term corrected GGA (PBE+U) and the screened hybrid functional HSE06. Our simulations are compared with the available experimental measurements. It is shown that the geometrical parameters computed with the HSE06 functional are the most accurate with respect to experiments, although PBE and PBE+U provide reliable results. Our results demonstrate that PBE gives reasonable estimations of the bandgaps and adding a U correction allows us to obtain bandgap values in very good agreement with respect to the experimental data. Surprisingly, the HSE06 functional is found to dramatically overestimate the band gaps of those vanadium oxides and therefore is not suitable to study these materials.
 Please wait while we load your content...
Please wait while we load your content...

