
Study of the influence of intermolecular interaction on classical and reverse substituent effects in para-substituted phenylboranes†‡
 *a
and
*a
and
Abstract
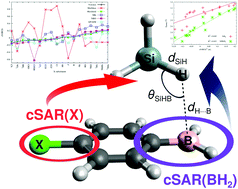
Using two families of XC6H4BH2⋯SiH4 and XC6H4BH2⋯SiMeH3 dimers where X represents 18 different substituents featuring a wide range of electronic properties, from significantly electron-withdrawing to significantly electron-donating, correlations between the cSAR(X) and cSAR(BH2) values, determined using the Mulliken, Hirshfeld, Merz-Kolmann, NBO, QTAIM, and Voronoi atomic charges, and various parameters are investigated. It is shown that the cSAR values obtained by means of the Hirshfeld atomic charges method match the quality obtained by the Voronoi method, while the Mulliken atomic charges method should definitely not be used for this purpose. For the first time, we notice that the quality of the correlation between cSAR values and parameters characterizing the strength of an intermolecular interaction depends to a large extent on the locality of the correlated parameter. Therefore, quite good linear correlations occur for the local distance H⋯B and the angle SiHB, while the correlations are poor for non-local interaction and binding energies. Moreover, cSAR values are strongly correlated with the parameters characterizing an intermolecular interaction only if this interaction has got a dominant influence on both the dimer structure and its overall binding effect. Thus relationships involving cSAR cannot always be used to predict the values of other properties, as the local or non-local character of the latter must be taken into account. Another important issue discussed in the article is the influence of the intermolecular interaction on the reverse substituent effect. It is shown that an intermolecular interaction can have a considerable influence on the electronic properties of the BH2 group, especially if X is a highly electron-withdrawing substituent. Most importantly, in contrast to the BH2 group itself, an intermolecular interaction reduces slightly the influence of the substituent effect. Although the influence on reverse substituent effect is small, the X substituents gain more electron charge. It has been shown that the presence of the BH2 group leads to both a significant reduction in the aromaticity of the benzene ring in XC6H4BH2 compared to XC6H5, as well as to its greater diversity in the former of these molecules. On the other hand, the influence of intermolecular interaction with either SiH4 or SiMeH3 on the aromaticity of the benzene ring in XC6H4BH2 is negligible.
 Please wait while we load your content...
Please wait while we load your content...

