
Whole-genome sequence analysis and homology modelling of the main protease and non-structural protein 3 of SARS-CoV-2 reveal an aza-peptide and a lead inhibitor with possible antiviral properties†
 *a
*a
Abstract
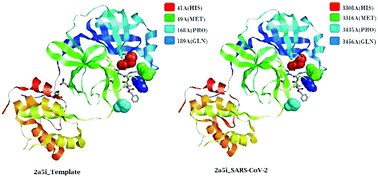
Viruses belonging to the family Coronaviridae consist of virulent pathogens that have a zoonotic property. Severe acute respiratory syndrome coronaviruses (SARS-CoVs) and Middle East respiratory syndrome coronaviruses (MERS-CoVs) of this family have emerged before and SARS-CoV-2 has emerged now globally. The characterization of spike glycoproteins, polyproteins and other viral proteins from viruses is important for antiviral drug development. Homology modelling of these proteins with known templates offers the opportunity to discover ligand-binding sites and explore the possible antiviral properties of these protein–ligand complexes. In this study, we performed a complete bioinformatic analysis, sequence alignment, comparison of multiple sequences and modelling of the SARS-CoV-2 whole-genome sequences, the spike protein and the polyproteins for homology with known proteins. We also analysed binding sites in these models for possible binding with ligands that exhibit antiviral properties. Our results indicated that the sequence of the polyprotein isolate SARS-CoV-2_HKU-SZ-001_2020 showed 98.94 percent identity to SARS-coronavirus NSP12 bound to NSP7 and NSP8 co-factors. The results also indicated that a part of the viral genome (residues 3268–3573 in Frame 2 with 306 amino acids) of the SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank Accession Number MN908947.3) when modelled with template 2a5i of the PDB database showed 96 percent identity to a 3C-like peptidase of SARS-CoVs, which has the ability to bind with an aza-peptide epoxide (APE) known for the irreversible inhibition of SARS-CoV main peptidase. A docking profile with 9 different conformations of the ligand with the protein model using Autodock Vina showed an affinity of −7.1 kcal mol−1. This region was conserved in 831 genomes of SARS-CoV-2. The part of the genome (residues 1568–1882 in Frame 2 with 315 amino acids) when modelled with template 3e9s of the PDB database showed 82 percent identity to a papain-like protease/deubiquitinase, which when complexed with ligand GRL0617 acts as an inhibitor and can block SARS-CoV replication. A docking profile with 9 different conformations of the ligand with the protein model using Autodock Vina showed an affinity of −7.9 kcal mol−1. This region was conserved in 831 genomes of SARS-CoV-2. It is possible that these ligands can be used as antivirals of SARS-CoV-2.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

